
Florent Laferrière et al. dans Nature Neuroscience
TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Laferrière F , Bezard E Picotti P Lashley T Polymenidou M et al. Nat Neurosci. 2019 Jan;22(1):65-77. doi: 10.1038/s41593-018-0294-y. Epub 2018 Dec 17.
Université de Bordeaux, Institut des Maladies Neurodégénératives, UMR 5293, Bordeaux, France. CNRS, Institut des Maladies Neurodégénératives, UMR 5293, Bordeaux, France. Laboratoire d’Erwan Bézard
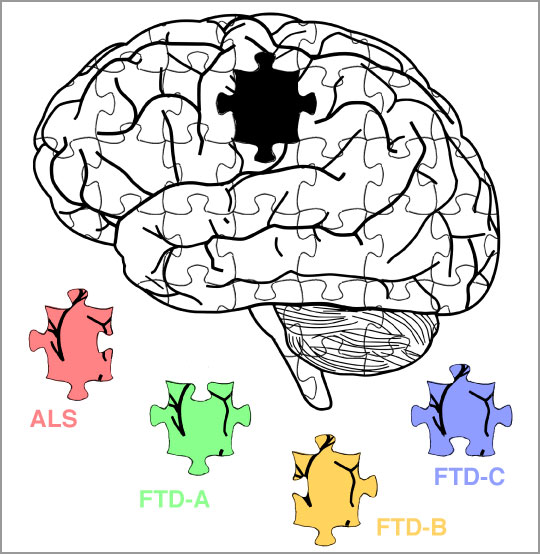
La sclérose latérale amyotrophique (maladie de Charcot), et la démence frontotemporale (maladie de Pick) sont des désordres neurodégénératifs liés à l’agrégation d’une protéine dans le cerveau des sujets atteints : TDP-43. Grâce au développement d’une nouvelle méthode d’extraction biochimique, des chercheurs du CNRS, de l’Inserm, et de l’Université de Bordeaux ont, en collaboration avec un laboratoire de l’Université de Zurich, identifié différents types d’assemblages pathologiques de cette protéine dans les cerveaux de sujets atteints de ces protéinopathies distinctes. Ces travaux sont publiés le 17 décembre dans la revue Nature Neuroscience.
La sclérose latérale amyotrophique (SLA, maladie de Charcot), et la démence frontotemporale (DFT, maladie de Pick) sont des maladies neurodégénératives d’issue fatale chez l’homme. La SLA se manifeste par une paralysie progressive et une atrophie musculaire, dues à la dégénérescence des neurones moteurs du cortex moteur et de la moelle épinière. La DFT, en revanche, se traduit par une démence consécutive à une importante mort neuronale dans le cortex frontal et temporal. Ces pathologies peuvent être d’origine génétique, mais la grande majorité des cas sont sporadiques. Outre des phénotypes extrêmement divergents, celles-ci présentent un spectre très large de causes, avec plusieurs dizaines de gènes identifiés, ainsi que des évolutions et des processus pathogéniques très diverses. La DFT présente elle-même des sous-types, différenciés par les signes cliniques ou anatomopathologiques.
Néanmoins, dans une très grande majorité des cas, un trait phénotypique majeur de ces pathologies est la présence d’inclusions cytoplasmiques composées (a minima) d’une protéine identifiée en 2006 : TDP-43 (Neumann et al, Science, 2006). Cette protéine lie l’ADN et l’ARN, et possède plusieurs rôles prépondérants dans le transport, l’épissage, et le métabolisme des ARN. Néanmoins, l’implication de TDP-43 dans la SLA et la DFT, son rôle précis dans la pathogénèse de ces maladies ainsi que les mécanismes aboutissants à une mort neuronale sont largement méconnus. (Photo: Florent Laferrière)
En développant une nouvelle méthode d’extraction des agrégats protéiques sur tissus cérébraux de sujets atteints de SLA ou DFT, les chercheurs ont pu isoler les assemblages pathologiques de TDP-43 associés à l’une ou l’autre maladie. Après purification, ils ont décrypté leurs propriétés physiques (taille, forme, densité, composition) et ont découvert que celles-ci diffèrent drastiquement suivant les protéinopathies à TDP-43, ce qui rappelle le phénomène de « souches » chez les virus ou les maladies à prions.
En inoculant ces agrégats protéiques distincts à des neurones en culture, les chercheurs ont déterminé qu’un type en particulier de ces assemblages protéiques était extrêmement toxique pour les cellules neuronales. De plus, ces agrégats les plus neurotoxiques sont observés spécifiquement chez les sujets atteints de DFT les plus agressives (rapidement létales), phénomène observé par exemple dans les différentes souches de prions.
Ces travaux fournissent donc une nouvelle méthode expérimentale d’extraction de ces agrégats de protéines pathologiques, et permettent une meilleure compréhension des mécanismes pathogéniques sous-jacents de ces maladies, dans l’optique d’ouvrir de nouvelles voies thérapeutiques.

Florent Laferrière
Institut des Maladies Neurodégénératives
CNRS UMR 5293 Université de Bordeaux,
Mise à jour: 12/05/21